

F127對三氧化鉬/氧化鋁結構及其催化氧化脫除苯并噻吩性能的影響

摘 要: 以PEO-PPO-PEO三嵌段共聚物 (F127, Ma=12 600) 為表面活性劑, 與鋁源AlCl3通過原位合成法制備了F127-MoO3/Al2O3催化劑, 并對催化劑進行了FT-IR、XRD、FE-SEM和N2吸附-脫附等表征。考察了不同反應條件對模型油催化氧化脫硫效果的影響。結果表明, 表面活性劑F127的加入促進了MoO3在Al2O3表面的高度分散, 提高了催化劑的比表面積、平均孔徑和孔容;對于10.0 m L模型油 (硫質量分數為400μg/g) , 在催化劑質量為0.06 g, 氧化劑用量為0.03 m L, 反應溫度為70℃, 反應時間為23 min時, 苯并噻吩 (BT) 的脫除率接近100%, BT被氧化成BTO2;催化劑重復使用5次后, 仍具有較高活性。
燃料油中的含硫化合物燃燒轉化為SOx是大氣污染和酸雨形成的重要來源之一, 而且會對一些控制和處理污染物的設備構成腐蝕破壞[1-2]。為了減少污染, 世界各國制定了更加嚴格的環保法規, 我國2018年將全面實施國五排放標準, 硫質量分數指標限值為10μg/g, 因此生產低硫甚至無硫燃料油成為必然的趨勢[3]。具有反應條件溫和, 不消耗大量氫氣, 投資少, 且對加氫脫硫 (HDS) 難以脫除的苯并噻吩及其衍生物有很好的脫除效果的氧化脫硫 (ODS) 工藝成為深度脫硫技術研究的熱點[4-5]。
對于ODS工藝, 研究的重點是開發出高活性、高穩定性且成本低的氧化脫硫催化劑。負載型鉬基催化劑以其優越的氧化脫硫催化性能而受到廣泛關注[6-7]。常用的鉬基催化劑載體有活性炭、二氧化鈦、二氧化硅、氧化鋁等, 其中氧化鋁由于具有可調的孔徑、較高的熱穩定性、低廉的成本等成為工業和學術界研究的焦點[8]。筆者選用H2O2作為氧化劑, 以F127為表面活性劑, 采用沉淀法制備了F127-Mo O3/Al2O3催化劑, 并對其結構進行了表征, 以苯并噻吩 (BT) 正辛烷溶液為模型油, 研究了其催化模型油氧化脫硫性能。
1 實驗
1.1 材料與試劑
鉬酸銨 (分析純) 、氯化鋁 (分析純) 、雙氧水 (質量分數為30%, 分析純) 、氨水 (分析純) 、甲醇 (分析純) 、乙醇 (分析純) 、正辛烷 (化學純) , 國藥集團化學試劑有限公司生產;苯并噻吩 (BT, 分析純) , Aladdin公司生產;聚醚F127 (化學純) , 薩恩化學技術 (上海) 有限公司生產。
1.2 儀器與設備
傅里葉變換-紅外光譜儀 (FT-IR) , Bruker VERTEX 70型, 德國Bruker公司生產;X射線衍射儀 (XRD) , Xpert Pro型, Cu Kα輻射, 荷蘭Philips公司生產;場發射掃描電子顯微鏡 (FE-SEM) , Nova400 Nano型, 美國FEI公司生產;比表面積和孔隙率分析儀 (BET) , Quadrasorb SI型, 美國康塔儀器有限公司生產;氣相色譜儀 (HP-6890) , FID檢測器, HP-5毛細管柱, 美國Agilent公司生產;全譜直讀等離子體發射光譜議 (ICP-AES) , IRIS Advantage ER/S型, 美國Thermo Elemental公司生產;氣相色譜-質譜聯用儀 (GC-MS) , ISQ型, 美國Thermofisher公司生產。
1.3 催化劑的制備
室溫條件下, 將0.67 g鉬酸銨和3.17 g 三氯化鋁分別溶于5.0 m L去離子水中備用, 向三口燒瓶中依次加入水、乙醇和氨水[V (水) ∶V (乙醇) ∶V (氨水) =20∶10∶1], 在1 000 r/min的攪拌速率下加入0.38 g F127, 待其充分溶解后依次滴加三氯化鋁溶液和鉬酸銨溶液, 持續攪拌1 h后將其在室溫下陳化4 h, 經過濾洗滌后, 在120℃下干燥12 h, 最后置于管式爐中升溫至540℃焙燒12 h (升溫過程及焙燒第1 h氮氣保護) , 將焙燒后的催化劑標記為F127-Mo O3/Al2O3;為了考察表面活性劑F127對催化劑結構的影響, 不加F127用同樣方法進行合成得到對比催化劑, 標記為Mo O3/Al2O3。
1.4 氧化脫硫反應
量取1.02 m L BT溶解于1 000 m L的正辛烷中, 得到硫質量分數為400μg/g的BT模型油。稱取一定量的預先干燥過的催化劑放入圓底燒瓶中, 然后向其中加入10.0 m L BT模型油, 在冷凝回流和電磁攪拌條件下進行水浴恒溫加熱, 待模型油的溫度達到設定溫度后, 向其中加入一定量的H2O2, 進行催化氧化脫硫反應。待反應結束后, 迅速取出圓底燒瓶并置入冰水浴中冷卻, 待其充分冷卻后, 通過離心操作分離出催化劑和模型油。利用氣相色譜儀對上層有機相進行含硫檢測, 計算模型油的脫硫率。
2 結果與討論
2.1 催化劑的表征
Mo O3、Mo O3/Al2O3和F127-Mo O3/Al2O3的紅外光譜如圖1所示。由圖1中譜線1可以看出, 1000cm以下的峰歸屬為鉬氧物種特征峰, 其中988 cm處的吸收峰為MoO雙鍵的伸縮振動吸收峰, 878、821 cm和634 cm處的譜峰為Mo—O—Mo鍵的伸縮振動吸收峰[9]。由圖1中譜線2和譜線3可以看出, 鉬氧物種的出峰被載體氧化鋁的Al—O鍵在870 cm附近的伸縮振動峰和該鍵在650cm附近的彎曲振動峰覆蓋, 表明MoO3在載體Al2O3表面得到較好分散[10]。
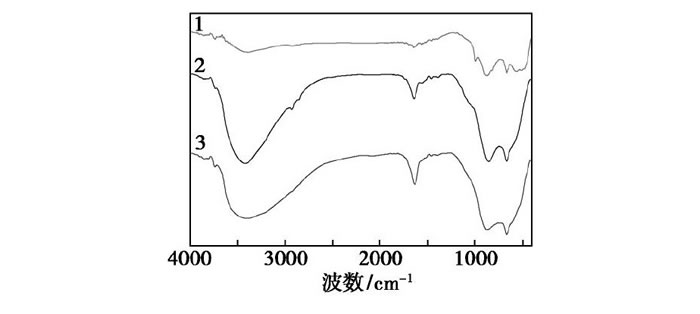
圖1 催化劑的FT-IR圖譜
1—Mo O3;2—Mo O3/Al2O3;3—F127-Mo O3/Al2O3
鉬酸銨、Mo O3/Al2O3和F127-Mo O3/Al2O3的XRD圖譜如圖2所示。由圖2可以看出, 對于540℃焙燒后的鉬酸銨, 在2θ=13、23、25、27、34、39、46、50、53、60°等位置檢測到了Mo O3的特征衍射峰[11]。催化劑Mo O3/Al2O3也出現了MoO3結構單元的特征衍射峰, 說明催化劑Mo O3/Al2O3中含有晶態MoO3;而對于F127-Mo O3/Al2O3, Mo O3結構單元的特征衍射峰并未出現, 活性中心MoO3在載體Al2O3上分散性較Mo O3/Al2O3催化劑更好, 催化劑總體表現為無定型結構。F127-Mo O3/Al2O3中, 鉬酸銨的活性中心MoO3在Al2O3載體高度分散, 這些結構特征有利于提高活性中心Mo O3與BT接觸幾率, 充分地發揮MoO3的催化活性, 提高催化劑對BT模型油的脫除率[12]。
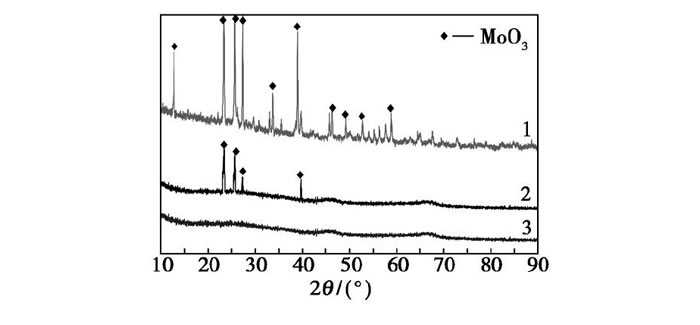
圖2 催化劑的XRD圖譜
1—鉬酸銨;2—Mo O3/Al2O3;3—F127-Mo O3/Al2O3
Mo O3/Al2O3和F127-Mo O3/Al2O3的掃描電鏡圖如圖3所示。從圖3中可以看出, Mo O3/Al2O3催化劑整體孔隙較少, 表面光滑;F127-Mo O3/Al2O3催化劑整體蓬松多孔, 主要是由于表面活性劑F127的加入使得催化劑在高溫焙燒過程中有較多的氣體逸出, 在催化劑內部和表面形成大量的孔洞, 蓬松多孔的結構更有利于催化劑對BT的吸附, 進而對其進行催化氧化, 達到高效脫除BT的目的[13]。

圖3 催化劑的FE-SEM圖
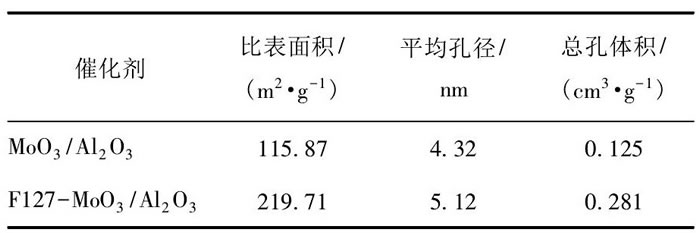
F127-Mo O3/Al2O3的吸附-脫附等溫線和孔徑分布情況如圖4所示。根據IUPAC的分類標準, F127-Mo O3/Al2O3的吸附脫附等溫線符合Ⅲ型吸附等溫線[14]。F127-Mo O3/Al2O3的孔徑分布基本全部集中在4.00~30.00 nm之間。Mo O3/Al2O3和F127-Mo O3/Al2O3的孔隙結構參數如表1所示。由表1可以看出, F127-Mo O3/Al2O3的平均孔徑為5.12 nm, 比表面積為219.71 m/g, 孔容為0.281 cm/g, 均高于催化劑Mo O3/Al2O3。表面活性劑F127在催化劑形成過程中起到有效的擴孔作用, 使得催化劑的比表面積、總孔體積和平均孔徑均增加。而較大的比表面積和孔容以及合適的孔徑分布更有利于活性中心Mo O3顯露在催化劑的表面, 顯著增加催化氧化過程中BT與催化劑活性中心的接觸幾率, 提高催化劑脫硫效率。
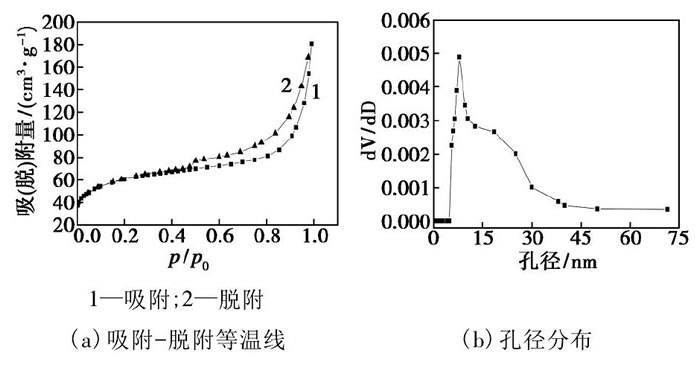
圖4 F127-Mo O3/Al2O3的吸附-脫附等溫線和孔徑分布
表1 催化劑的孔隙結構參數
2.2 模型油的氧化脫硫
2.2.1 催化劑質量對脫硫率的影響
在H2O2的用量為0.03 m L, 反應溫度為70℃, 反應時間為25 min時, 分別研究了F127-Mo O3/Al2O3和Mo O3/Al2O3的質量對BT模型油氧化脫硫率的影響, 結果如圖5所示。從圖5中可以看出, 隨著催化劑質量的增加, BT模型油的脫硫率逐漸升高, 在相同的催化劑質量條件下, 催化劑F127-Mo O3/Al2O3對BT模型油的脫除率明顯高于催化劑Mo O3/Al2O3。當催化劑F127-Mo O3/Al2O3的質量為0.06 g時, BT的脫除率接近100%, 因此, 選擇F127-Mo O3/Al2O3的質量0.06 g作為催化氧化脫除BT模型油的最佳催化劑質量。
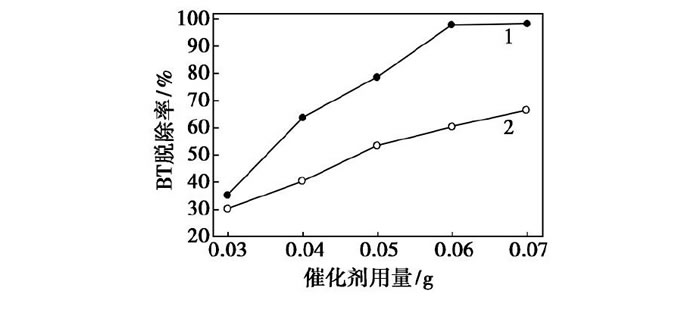
圖5 催化劑質量對BT脫除率的影響
1—F127-Mo O3/Al2O3;2—Mo O3/Al2O3
2.2.2 氧化劑用量對脫硫率的影響
在催化劑F127-Mo O3/Al2O3或Mo O3/Al2O3質量為0.06 g, 反應溫度為70℃, 反應時間為25 min時, 考察了雙氧水用量對BT模型油氧化脫硫率的影響, 結果如圖6所示。由圖6可以看出, BT模型油的脫硫率隨著H2O2用量的增加呈現出先增加后降低的趨勢, H2O2用量增加會增加反應中間體過氧金屬化合物的生成量, 有利于加快反應速率;進一步提高H2O2用量時, 脫硫率出現下降, H2O2用量增加的同時也會給反應體系帶來更多的H2O, 制備的F127-Mo O3/Al2O3和Mo O3/Al2O3都具有親水性能, 更傾向于向反應體系生成的水相中轉移, 降低其在模型油中的分散性, 導致催化劑活性降低[15]。對于催化劑F127-Mo O3/Al2O3, 當H2O2的用量為0.03 m L時, 其催化氧化脫除BT模型油的脫硫率最佳, 接近100%。所以F127-Mo O3/Al2O3的最佳氧化劑用量為0.03 m L。
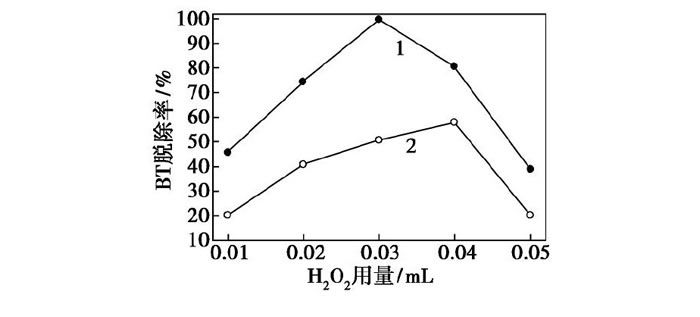
圖6 H2O2用量對BT脫除率的影響
1—F127-Mo O3/Al2O3;2—Mo O3/Al2O3
2.2.3 反應溫度對脫硫率的影響
在催化劑F127-Mo O3/Al2O3或Mo O3/Al2O3質量為0.06 g, H2O2用量為0.03 m L, 反應時間為25 min時, 考察反應溫度對BT模型油氧化脫硫率的影響, 結果如圖7所示。從圖7中曲線1可以看出, 隨著反應溫度的升高, 催化劑F127-Mo O3/Al2O3對BT的脫除率逐漸升高, 在反應溫度為70℃時, BT的轉化率已經達到97.30%;在反應溫度為80℃時, BT的轉化率接近100%。從圖7中曲線2可以看出, 隨著反應溫度的升高, 催化劑、Mo O3/Al2O3對BT的脫除率逐漸升高, 但是相同條件下的脫硫率明顯低于催化劑F127-Mo O3/Al2O3。然而, 反應溫度為80℃時, 不僅會使脫硫操作成本升高, 還會加快H2O2的分解速率。因此, F127-Mo O3/Al2O3脫除BT模型油的最佳反應溫度為70℃。
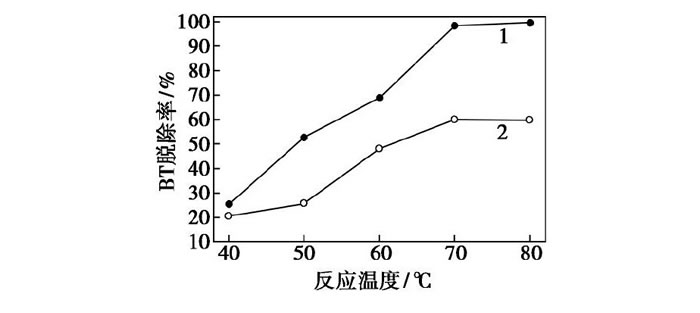
圖7 反應溫度對BT脫除率的影響
1—F127-Mo O3/Al2O3;2—Mo O3/Al2O3
2.2.4 反應時間對脫硫率的影響
在催化劑F127-Mo O3/Al2O3或Mo O3/Al2O3質量為0.06 g, H2O2用量為0.03 m L, 反應溫度為70℃時, 考察反應時間對BT模型油氧化脫硫率的影響, 結果如圖8所示。由圖8中曲線1可以看出, 隨著反應時間的增加, BT模型油的脫硫率逐漸升高, 在反應時間為23 min時, 脫硫率接近100%。而從圖8中曲線2中可以看出, 反應進行到23 min時, BT的脫除率僅為63.50%。在相同反應條件下, 催化劑F127-Mo O3/Al2O3的催化活性均優于催化劑Mo O3/Al2O3。
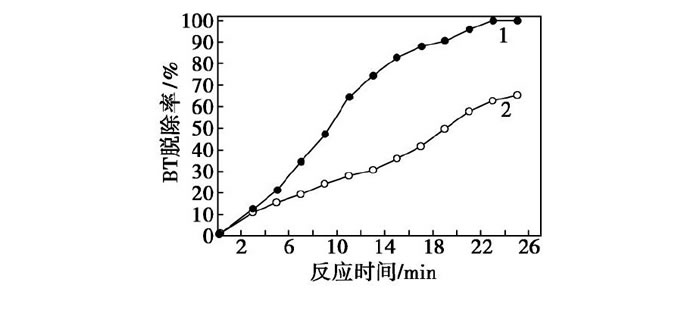
圖8 反應時間對BT脫除率的影響
1—F127-Mo O3/Al2O3;2—Mo O3/Al2O3
2.3 BT氧化產物的分離與分析
在催化劑F127-Mo O3/Al2O3催化氧化脫除BT模型油的最佳反應條件下進行脫硫實驗, 實驗結束后用氣相色譜儀檢測模型油中硫的質量分數, 結果并未檢測到含硫化合物。將進行脫硫反應后的催化劑F127-Mo O3/Al2O3用甲醇洗滌30 min, 然后將甲醇洗滌液和催化劑離心分離, 并用氣相色譜-質譜聯用儀對甲醇溶液進行檢測, 氧化產物質譜圖如圖9所示。通過與標準NIST譜庫對比可以得出, 圖9中m/z=166.08所對應的峰歸屬為苯并噻吩砜 (BTO2) 的分子離子峰, BT幾乎全部被氧化成BTO2, 并且BTO2被催化劑表面吸附, 反應后的模型油中幾乎不含BT和BTO2。
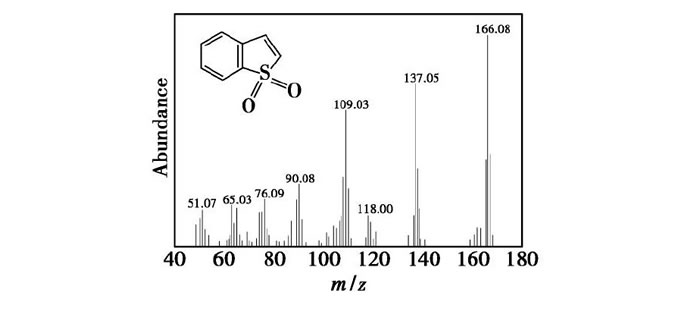
圖9 BT氧化產物MS圖譜
2.4 催化劑重復使用性
在最佳反應條件下, 利用催化劑F127-Mo O3/Al2O3進行催化氧化脫硫反應, 反應結束后將催化劑F127-Mo O3/Al2O3與模型油分離后回收, 并用甲醇浸泡10 min后過濾, 在105℃的干燥箱內干燥再生。催化劑再生后在最佳實驗條件下進行脫硫反應, 并進行重復性實驗, 結果如表2所示。從表2中可以看出, 當催化劑重復使用5次后, BT的轉化率可以達到93.80%, 仍具有較好的脫硫活性, 表明催化劑F127-Mo O3/Al2O3具有良好的重復使用性能。對脫硫前后催化劑F127-Mo O3/Al2O3中的Mo的質量分數進行ICP-AES檢測, 檢測結果為Mo與Al2O3的質量比由重復使用5次前的30.72%降低為28.91%, 可見在反應過程中F127-Mo O3/Al2O3中的Mo O3有少量發生溶脫, 而且強極性的氧化產物BTO2在催化劑的使用過程中也會有所殘留, 因此, 重復使用時催化劑F127-Mo O3/Al2O3對BT模型油的脫除率有所降低[16]。
表2 催化劑的重復使用性能研究

參考文獻
[1]Tao X, Zhou Y, Wei Q, et al.Inhibiting effects of nitrogen compounds on deep hydrodesulfurization of straight-run gas oil over a Ni W/Al2O3catalyst[J].Fuel, 2017, 188:401-407.
[2]Zaid H F M, Chong F K, Mutalib M I A.Extractive deep desulfurization of diesel using choline chloride-glycerol eutectic-based ionic liquid as a green solvent[J].Fuel, 2017, 192:10-17.
[3]宋紅艷, 何靜, 李春喜.燃料油深度脫硫的技術策略及研究進展[J].石油化工, 2015, 44 (3) :279-286.
[4]Hajjar Z, Kazemeini M, Rashidi A, et al.Graphene based catalysts for deep hydrodesulfurization of naphtha and diesel fuels:A physiochemical study[J].Fuel, 2016, 165:468-476.
[5]Dedual G, Macdonald M, Alshareef A, et al.Requirements for effective photocatalytic oxidative desulfurization of a thiophenecontaining solution using Ti O2[J].Journal of Environmental Chemical Engineering, 2014, 2 (4) :1947-1955.
[6]劉靜, 劉東, 杜輝, 等.鉬基改性催化劑的制備及其催化氧化脫硫性能研究[J].石油煉制與化工, 2016, 47 (3) :42-47.
[7]García-Gutiérrez J, Fuentes G, Hernndez-Tern M E, et al.Ultradeep oxidative desulfurization of diesel fuel by the Mo/Al2O3-H2O2, system:The effect of system parameters on catalytic activity[J].Applied Catalysis A General, 2008, 334 (1-2) :366-373.
[8]García-Gutiérrez J, Fuentes G, Hernndez-Tern M, et al.Ultra-deep oxidative desulfurization of diesel fuel with H2O2, catalyzed under mild conditions by polymolybdates supported on Al2O3[J].Applied Catalysis A General, 2006, 305 (1) :15-20.
[9]寧文生, 卞國柱.表面鉬物種的紅外光譜研究[J].浙江工業大學學報, 2001, 29 (3) :213-216.
[10]劉志軍, 陸江銀.Mo O3/γ-Al2O3催化劑上正丁烷催化脫氫制正丁烯[J].化工生產與技術, 2009, 16 (2) :20-23.
[11]朱伯仲, 林鈺, 尚雪亞, 等.鉬酸銨的熱分解機理研究[J].蘭州大學學報 (自科版) , 1997 (3) :72-76.
[12]田永勝, 王光輝, 趙磊, 等.聚乙二醇對HPMo/Si O2結構及其催化氧化脫除二苯并噻吩性能的影響[J].石油煉制與化工, 2015, 46 (10) :50-55.
[13]田永勝, 王光輝, 趙磊, 等.PEG-HPMo/Si O2介孔材料的制備及其催化氧化脫除模型油中苯并噻吩的研究[J].材料導報, 2015 (16) :22-26.
 同類文章排行
同類文章排行 最新資訊文章
最新資訊文章
